AlphaFold Uncovered: How AI Maps the Building Blocks of Life





Introduction
AlphaFold is a revolutionary AI system developed by DeepMind that predicts 3D structures of proteins and protein complexes from amino acid sequences. Its evolution from AlphaFold 1 to AlphaFold 3 has redefined the field of structural biology, enabling researchers to simulate molecular interactions with unprecedented accuracy. In this blog post, we will trace the evolution of AlphaFold, explore the AI architectures and datasets that power it, and demonstrate its use in modeling protein-RNA-DNA interactions, particularly with a biologically relevant example.
The Evolution of AlphaFold: From Single Proteins to Complex Interactions
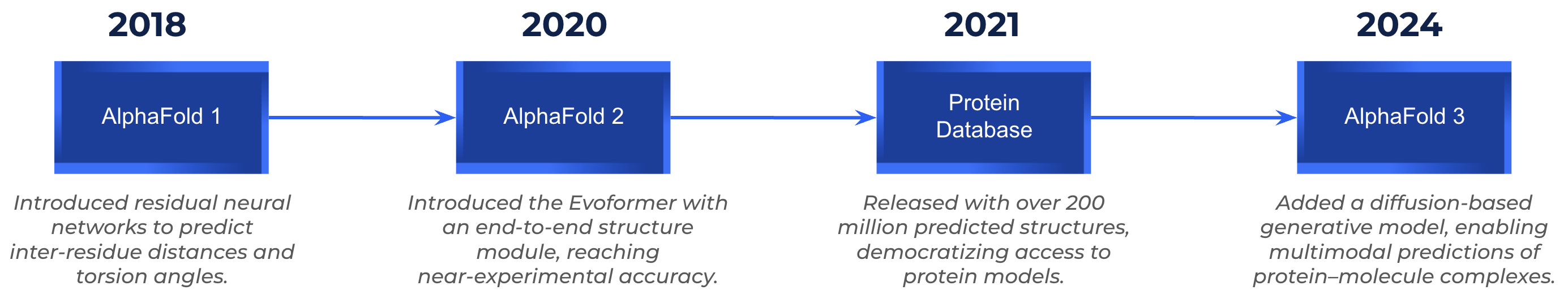
AlphaFold 1 (2018): The Breakthrough at CASP13
Before AlphaFold, determining a protein’s 3D structure relied on experimental techniques such as X-ray crystallography or cryo-electron microscopy, which were highly accurate but also slow and expensive. To benchmark computational alternatives, the CASP competition (Critical Assessment of Structure Prediction) was created, challenging teams to predict unseen protein structures that would later be compared against experimental data. By 2018, despite decades of research, computational methods still struggled to rival experimental precision. That changed when DeepMind entered the scene.
The first version of AlphaFold made headlines in 2018, when a team of AI researchers from DeepMind, not traditional structural biologists, won the CASP13 challenge on their first attempt. AlphaFold 1 used a two-part pipeline:
- A gradient descent-based energy minimization process to assemble 3D structures from the predicted geometries.
- A deep residual convolutional neural network (CNN) that predicted inter-residue distances and torsion angles from evolutionary data.
%2010.40.50%E2%80%AFa.%C2%A0m..png)
Its key innovation was leveraging co-evolutionary information extracted from multiple sequence alignments (MSAs), which are sets of homologous protein sequences aligned across species. By using pre-computed evolutionary correlations from MSAs, where certain amino acid positions tend to mutate together across evolution, AlphaFold 1 learned to map these patterns into 3D spatial proximity between residues. The model produced a distance map (or distogram), a 2D matrix representing the most probable distances between every pair of residues, which served as the foundation for reconstructing the full 3D fold.
%2010.43.20%E2%80%AFa.%C2%A0m..png)
Although its predictions were not always atomic-level, they significantly outperformed traditional homology models, especially in the free-modeling category. AlphaFold 1 marked a turning point, showing that a complex biological problem, long dominated by physics-based modeling, could be effectively reframed as a machine learning task.
AlphaFold 2 (2020-2021): A Quantum Leap in Prediction Accuracy
The second generation of AlphaFold marked a radical shift in how protein structures were predicted. While AlphaFold 1 relied on pre-computed features such as evolutionary correlations, AlphaFold 2 replaced this multi-step pipeline with a single end-to-end deep learning system that learns directly from sequence data. Its architecture was built around two key components:
- Evoformer: a transformer-based network that simultaneously processes information from multiple sequence alignments (MSAs) and pairwise residue relationships. By building internal embeddings for both the MSA and the residue pairs, the Evoformer captures evolutionary and spatial relationships directly, without the need for pre-calculated covariance matrices.
- Structure Module: a network that takes the refined representations from the Evoformer and predicts 3D atomic coordinates directly, refining them iteratively through a process known as recycling, where each prediction pass improves upon the previous one.
This transformer design fundamentally changed how AlphaFold processed biological information. Instead of handcrafted features, the model uses self-attention mechanisms, the same concept behind natural language models like BERT, to capture long range dependencies between amino acids, treating the protein sequence like a “sentence” where context matters for each residue. Improvements included:
- End-to-end learning from raw sequence to final 3D structure.
- Transformer-based attention that integrates both sequence and spatial information.
- Recycling of predictions to improve accuracy over multiple passes.
AlphaFold 2 won CASP14 by a wide margin, achieving near-experimental accuracy (GDT > 90) on two-thirds of targets. Beyond the competition, its open source release and the creation of the AlphaFold Protein Structure Database, containing over 200 million predicted structures, democratized access to protein structure data worldwide.
%2010.46.50%E2%80%AFa.%C2%A0m..png)
AlphaFold 3 (2024): Modeling the Molecular Machinery of Life
AlphaFold 3, released in 2024, took a major step forward by enabling the prediction of multi-component complexes, including:
- Protein-protein assemblies
- Protein-DNA/RNA interactions
- Protein-ligand and ion binding sites
Unlike AlphaFold 2, which required separate models for multimers or additional scripts, AlphaFold 3 uses a unified architecture to model all components simultaneously.
While the inference code of AlphaFold 3 is publicly available on DeepMind’s GitHub, the model weights are distributed separately under a non-commercial academic license and must be requested from DeepMind. This makes AlphaFold 3 a research-only release rather than a fully open-source model, with most users accessing it through the official AlphaFold Server.
The AI Learning Behind AlphaFold 3
Core Architecture: From Evoformer to Diffusion Models
AlphaFold 3 retains the Evoformer module for handling input sequences but replaces the structure module with a diffusion-based generative model. This new approach starts from a random cloud of atoms and iteratively refines them into a stable 3D structure, similar to how image diffusion models like Stable Diffusion generate coherent images from noise.
%2010.50.01%E2%80%AFa.%C2%A0m..png)
Key features of AlphaFold 3’s architecture:
- Multimodal input support: amino acids, nucleotides, ligands, ions.
- Joint structural prediction: all chains and cofactors predicted together.
- Template-free inference: predictions made directly from sequence, optionally guided by templates.
Datasets Used in Training
AlphaFold 3 was trained on a broad collection of biological data, expanding both the sequence and structural diversity used in previous versions.
Sequence and Evolutionary Databases:
The model leveraged several large public datasets, including UniProt, BFD, and MGnify. Additional sources included Uniclust30, RFam, RNAcentral, and the Nucleotide Database.
Structural Databases:
It was trained and evaluated using experimental 3D data from the Protein Data Bank (PDB), as well as nucleic-acid structures from the Nucleotide Database and ligand information from the Chemical Components Dictionary (CCD).
Data Format and Preprocessing:
Input features include multiple sequence alignments (MSAs) and templates, although MSA processing is less central than in AlphaFold 2. Templates are retrieved via a search module, and inputs are embedded into pairwise representations that are processed by the Pairformer, which replaces the AlphaFold 2 Evoformer.
Training Objectives:
AlphaFold 3 was trained through a generative diffusion framework, combining:
- Supervised learning: predicting “denoised” atomic coordinates from corrupted inputs.
- Cross-distillation: augmenting data with pseudo-structures from AlphaFold-Multimer v2.3 to mitigate hallucination.
- Generative learning on evolutionary data: allowing the model to capture both global folds and local stereochemistry across varying noise levels.
Case Study: Modeling the CRISPR-Cas9 Complex with AlphaFold 3
Background
AlphaFold 3 not only predicts the three dimensional folding of proteins but also allows researchers to explore how different types of biomolecules interact with others, including proteins, DNA, RNA, and small ligands. To illustrate this, we performed two example experiments using the AlphaFold Server, modeling human hemoglobin, a well known multi protein complex, and the CRISPR-Cas9 system, a protein-RNA-DNA assembly central to modern gene editing.
Before diving into the results, it is worth briefly explaining how the server works and how to interpret its confidence metrics.
How predictions are made in AlphaFold Server
The AlphaFold Server provides an intuitive interface for modeling molecules directly from their sequences.
Each entity (protein, DNA, RNA, ligand, or ion) can be added as a separate chain by pasting its FASTA sequence. Once all entities are configured, the server performs inference using the AlphaFold 3 model, which predicts the 3D coordinates of every component simultaneously.
%2010.53.36%E2%80%AFa.%C2%A0m..png)
To reproduce this setup, the sequences used correspond to the experimental structure PDB 5F9R, which contains the Cas9 protein, guide RNA, and target DNA duplex.
Full FASTA files for each entity can be copied directly from the RCSB PDB entry 5F9R via the FASTA display page.
The results include a predicted structure, numerical confidence scores, and color coded visualizations that help assess model reliability at a glance.
%2010.54.49%E2%80%AFa.%C2%A0m..png)
The example shows a 3D structure colored by plDDT alongside a Predicted Aligned Error (PAE) matrix. The PAE map estimates how accurately different regions or chains are positioned relative to one another, dark green indicates lower alignment error (higher confidence), and lighter shades indicate greater uncertainty in relative positioning.
After generating a prediction, AlphaFold 3 reports three main confidence indicators, displayed at the top of each result:
- plDDT (Predicted Local Distance Difference Test) measures local confidence for each residue. Color scale: blue = very high (> 90), light blue = confident (70-90), yellow = low (50-70), orange = very low (< 50). These colors are applied directly to the 3D model to highlight rigid versus flexible regions.
- pTM (Predicted Template Modeling score) evaluates the overall quality of the fold within a single chain.
- ipTM (Inter-chain Predicted TM score) quantifies how confidently AlphaFold 3 predicts interactions between different chains, such as between protein subunits or between a protein and a nucleic acid.
Case 1: Human Hemoglobin -From Subunits to a Functional Complex
Hemoglobin is one of the most emblematic protein assemblies in biology. It is composed of four subunits, two alpha (HBA) and two beta (HBB), that together form the tetrameric complex responsible for oxygen transport in red blood cells. To explore how AlphaFold 3 handles both individual proteins and multimeric structures, we first modeled each subunit separately and then the complete hemoglobin complex containing 2×HBA and 2×HBB.
To reproduce this experiment, the hemoglobin sequences were obtained from the experimental structure PDB 1A3N, available on the RCSB PDB entry 1A3N via the FASTA link.
%2011.26.39%E2%80%AFa.%C2%A0m..png)
Across all three predictions, AlphaFold 3 displayed consistently high confidence. Both HBA and HBB subunits showed similar plDDT distributions, dominated by blue and light blue regions that indicate high local reliability, with only small flexible termini in yellow or orange. The pTM scores were also high (0.85 for HBA and 0.88 for HBB) confirming accurate monomeric folds. Since these subunits were modeled individually, ipTM scores are not applicable (no inter-chain interactions are present).
When the full tetramer was modeled, AlphaFold 3 produced an even more confident result, with pTM = 0.89 and ipTM = 0.86, while the predicted structure appeared almost entirely blue. This reflects the model’s higher certainty in predicting the quaternary arrangement compared to isolated subunits.
In biological terms, this makes sense, when alpha and beta chains combine, they form a rigid, evolutionarily conserved complex that AlphaFold can recognize from its training data, reducing uncertainty and increasing overall confidence.
The resulting structure closely resembles the canonical hemoglobin fold, capturing its symmetric organization and internal stability.
%2011.27.57%E2%80%AFa.%C2%A0m..png)
The visual agreement between the crystallographic structure and the AlphaFold 3 prediction is remarkable. The model accurately reproduces the relative orientation of all four subunits and the central cavity that characterizes hemoglobin’s oxygen-binding form. The alignment between the two representations highlights how AlphaFold 3 not only captures atomic-level details but also the global geometry that defines the functional tetramer. This close match reinforces AlphaFold 3’s reliability in predicting multi-protein assemblies that have well conserved architectures across species.
Case 2: CRISPR-Cas9: Predicting a Protein-RNA-DNA Complex
One of the most transformative innovations introduced by AlphaFold 3 is its ability to model not only proteins, but also their interactions with other types of biomolecules, including DNA, RNA, ligands, and ions. This represents a major leap forward compared to previous versions, which were limited to protein-protein complexes. The model now captures how proteins engage with nucleic acids, a key aspect of many biological processes such as transcription, replication, and genome editing.
A clear example of this new multimodal capability is the CRISPR-Cas9 system, a molecular complex that combines a Cas9 endonuclease protein, a guide RNA (gRNA), and a double-stranded DNA target. Cas9 uses the RNA molecule as a guide to locate and cut a complementary DNA sequence, enabling precise gene editing.
To test AlphaFold 3’s ability to predict such intricate assemblies, we modeled these three entities simultaneously in the AlphaFold Server:
- The Cas9 protein
- The guide RNA
- The double-stranded DNA containing the target site
(The setup of these four entities was shown earlier in Figure 6.)
%2011.29.06%E2%80%AFa.%C2%A0m..png)
The similarity between the experimental and predicted structures is striking, especially given the multimolecular nature of the system. AlphaFold 3 accurately recapitulates the global geometry of the Cas9-RNA-DNA assembly, capturing the positioning of the guide RNA within the protein’s catalytic cleft and the bending of the DNA duplex around it.
As seen in the plDDT-colored model (right panel), most of the structure appears in blue and light blue, reflecting strong local confidence across Cas9 and the RNA-DNA interface. Only small regions appear in yellow or orange, mainly within one of the DNA strands (pink), which also corresponds to the area where the greatest deviation from the crystallographic reference is observed.
These results highlight how AlphaFold 3 successfully extends beyond protein folding to faithfully reconstruct functional macromolecular interactions. By integrating proteins and nucleic acids within a single predictive framework, it enables a new level of insight into molecular mechanisms of genome editing and other complex biological systems.
Biotech Applications of AlphaFold
AlphaFold is redefining how biotech teams approach molecular design and discovery. Its ability to predict not just individual protein structures, but also complex interactions with DNA, RNA, ligands, and other proteins, unlocks a wide range of real-world applications.
In drug discovery, AlphaFold accelerates the process of identifying how therapeutic molecules bind to their targets, streamlining lead design and reducing experimental overhead. When it comes to gene regulation or viral replication, the model helps visualize how proteins interact with RNA and DNA, bringing new insights into RNP assemblies and CRISPR mechanisms.
For synthetic biology, it enables the design of entirely new enzymes and multi-protein complexes, pushing the boundaries of what’s possible in metabolic engineering or industrial biocatalysis. In immunotherapy, AlphaFold plays a key role in guiding antibody-antigen modeling, revealing critical contact sites and helping optimize monoclonal antibody candidates.
And with tools like AlphaMissense, researchers can pair AlphaFold’s structural predictions with genomic variant data offering interpretations of mutations and their functional consequences.

Conclusion
From AlphaFold 1’s early convolutional approaches to AlphaFold 2’s transformer based architecture and now AlphaFold 3’s diffusion driven generative modeling, the progression of this system mirrors the evolution of modern AI itself. Each iteration has brought the scientific community closer to understanding how the language of sequences translates into the architecture of life.
What truly sets AlphaFold 3 apart is its expansion beyond proteins, its ability to predict interactions between proteins, nucleic acids, ligands, and ions within a single unified framework. This leap transforms the problem from predicting individual folds to modeling molecular assemblies and functional interactions, as demonstrated in our hemoglobin and CRISPR-Cas9 examples.
By accurately capturing not only atomic level details but also the dynamic interplay between biomolecules, AlphaFold 3 represents a shift from static structural prediction toward mechanistic understanding. It enables researchers to visualize, in silico, complexes that were once accessible only through years of experimental effort.
In this sense, AlphaFold 3 stands as a testament to how machine learning can transform our understanding of biology, bridging computation and experiment to uncover the fundamental principles that govern molecular life.
References
[1] Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., Qin, C., Žídek, A., Nelson, A. W. R., Bridgland, A., Penedones, H., Petersen, S., Simonyan, K., Crossan, S., Kohli, P., Jones, D. T., Silver, D., Kavukcuoglu, K., & Hassabis, D. (2020). Improved protein structure prediction using potentials from deep learning. Nature, 577, 706-710.
https://doi.org/10.1038/s41586-019-1923-7
(AlphaFold 1 — model used in CASP13, published in 2020.)
[2] Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., ... Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583-589.
https://doi.org/10.1038/s41586-021-03819-2
(AlphaFold 2 — CASP14-winning model, open-sourced in 2021.)
[3] Abramson, J., Jumper, J., Silver, D., Hassabis, D., & the DeepMind Team. (2024). AlphaFold 3 predicts the structure and interactions of all of life’s molecules.Nature, 630, 493-500.https://doi.org/10.1038/s41586-024-07487-w
(AlphaFold 3 — multimolecular model supporting proteins, RNA, DNA, and ligands.)


.png)