AlphaFold descubierto: cómo la IA mapea los componentes básicos de la vida





Introducción
AlphaFold es un revolucionario sistema de IA desarrollado por DeepMind que predice las estructuras tridimensionales de las proteínas y complejos proteicos a partir de secuencias de aminoácidos. Su evolución de AlphaFold 1 a AlphaFold 3 ha redefinido el campo de la biología estructural, permitiendo a los investigadores simular interacciones moleculares con una precisión sin precedentes. En esta entrada del blog, analizaremos la evolución de AlphaFold, exploraremos las arquitecturas de inteligencia artificial y los conjuntos de datos que lo impulsan y demostraremos su uso para modelar las interacciones entre proteínas, ARN y ADN, especialmente con un ejemplo biológicamente relevante.
La evolución de AlphaFold: de proteínas individuales a interacciones complejas
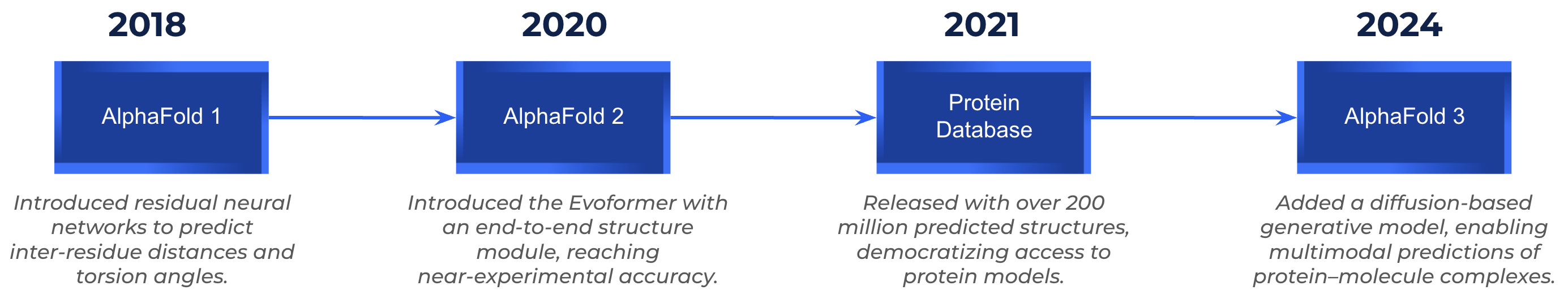
AlphaFold 1 (2018): el gran avance en CASP13
Antes de AlphaFold, la determinación de la estructura tridimensional de una proteína se basaba en técnicas experimentales como la cristalografía de rayos X o la microscopía crioelectrónica, que eran muy precisas pero también lentas y costosas. Para comparar las alternativas computacionales, se creó el concurso CASP (Critical Assessment of Structure Prediction, evaluación crítica de la predicción de estructuras), en el que los equipos tenían que predecir estructuras proteicas desconocidas para luego compararlas con datos experimentales. En 2018, a pesar de décadas de investigación, los métodos computacionales seguían teniendo dificultades para competir con la precisión experimental. Eso cambió cuando DeepMind entró en escena.
La primera versión de AlphaFold llegó a los titulares en 2018, cuando un equipo de investigadores de IA de DeepMind, no biólogos estructurales tradicionales, ganó el desafío CASP13 en su primer intento. AlphaFold 1 utilizó una canalización de dos partes:
- UN minimización de energía basada en gradiente descendente proceso para ensamblar estructuras 3D a partir de las geometrías predichas.
- Un profundo red neuronal convolucional residual (CNN) que predijo las distancias entre residuos y los ángulos de torsión a partir de datos evolutivos.
%2010.40.50%E2%80%AFa.%C2%A0m..png)
Su innovación clave fue aprovechar información coevolutiva extraída de alineaciones de secuencias múltiples (MSA), que son conjuntos de secuencias proteicas homólogas alineadas entre especies. AlphaFold 1 aprendió a mapear estos patrones para establecer una proximidad espacial tridimensional entre los residuos mediante el uso de correlaciones evolutivas precalculadas a partir de los MSA, en las que ciertas posiciones de los aminoácidos tienden a mutar juntas a lo largo de la evolución. El modelo produjo un mapa de distancias (o distograma), una matriz 2D que representa las distancias más probables entre cada par de residuos, que sirvió de base para reconstruir el pliegue tridimensional completo.
%2010.43.20%E2%80%AFa.%C2%A0m..png)
Aunque sus predicciones no siempre fueron de nivel atómico, superaron significativamente a los modelos de homología tradicionales, especialmente en la categoría de modelado libre. AlphaFold 1 marcó un punto de inflexión, pues demostró que un problema biológico complejo, dominado durante mucho tiempo por la modelización basada en la física, podía replantearse eficazmente como una tarea de aprendizaje automático.
AlphaFold 2 (2020-2021): un salto cualitativo en la precisión de las predicciones
La segunda generación de AlphaFold marcó un cambio radical en la forma en que se predijeron las estructuras de las proteínas. Mientras que AlphaFold 1 se basaba en características precalculadas, como las correlaciones evolutivas, AlphaFold 2 sustituyó este proceso de varios pasos por un un único sistema de aprendizaje profundo de extremo a extremo que aprende directamente de los datos de secuencia. Su arquitectura se construyó en torno a dos componentes clave:
- Evoformador: una red basada en transformadores que procesa simultáneamente la información de múltiples alineaciones de secuencias (MSA) y relaciones de residuos por pares. Al crear incrustaciones internas tanto para el MSA como para los pares de residuos, el Evoformer captura directamente las relaciones evolutivas y espaciales, sin necesidad de matrices de covarianzas precalculadas.
- Módulo de estructura: una red que toma las representaciones refinadas del Evoformer y predice directamente las coordenadas atómicas 3D, refinándolas iterativamente mediante un proceso conocido como reciclaje, en el que cada pasada de predicción mejora la anterior.
El diseño de este transformador cambió radicalmente la forma en que AlphaFold procesaba la información biológica. En lugar de características hechas a mano, el modelo usa mecanismos de autoatención, el mismo concepto detrás de los modelos de lenguaje natural como BERT, para capturar las dependencias de largo alcance entre los aminoácidos y tratar la secuencia de proteínas como una «oración» en la que el contexto es importante para cada residuo. Las mejoras incluyeron:
- Aprendizaje de principio a fin desde la secuencia sin procesar hasta la estructura 3D final.
- Atención basada en transformadores que integra información espacial y de secuencia.
- Reciclaje de predicciones para mejorar la precisión en varias pasadas.
AlphaFold 2 ganó el CASP14 por un amplio margen, lograr una precisión casi experimental (GDT > 90) sobre dos tercios de las metas. Más allá de la competencia, su lanzamiento de código abierto y la creación de la base de datos sobre la estructura de las proteínas AlphaFold, que contiene más de 200 millones de estructuras predichas, democratizaron el acceso a los datos sobre la estructura de las proteínas en todo el mundo.
%2010.46.50%E2%80%AFa.%C2%A0m..png)
AlphaFold 3 (2024): Modelando la maquinaria molecular de la vida
AlphaFold 3, lanzado en 2024, dio un gran paso adelante al permitir la predicción de complejos multicomponentes, que incluyen:
- Ensamblajes proteína-proteína
- Interacciones proteína-ADN/ARN
- Sitios de unión de proteínas, ligandos e iones
A diferencia de AlphaFold 2, que requería modelos separados para multímeros o scripts adicionales, AlphaFold 3 usa un arquitectura unificada para modelar todos los componentes simultáneamente.
Si bien el código de inferencia de AlphaFold 3 está disponible públicamente en el GitHub de DeepMind, las ponderaciones del modelo se distribuyen por separado bajo una licencia académica no comercial y deben solicitarse a DeepMind. Esto convierte a AlphaFold 3 en una versión exclusiva para fines de investigación y no en un modelo totalmente de código abierto, ya que la mayoría de los usuarios acceden a él a través de la versión oficial Servidor AlphaFold.
El aprendizaje de la IA detrás de AlphaFold 3
Arquitectura central: de Evoformer a Diffusion Models
AlphaFold 3 conserva el módulo Evoformer para gestionar las secuencias de entrada, pero reemplaza el módulo de estructura por un modelo generativo basado en la difusión. Este nuevo enfoque parte de una nube aleatoria de átomos y los refina de forma iterativa hasta obtener una estructura 3D estable, similar a la que utilizan los modelos de difusión de imágenes como Difusión estable generar imágenes coherentes a partir del ruido.
%2010.50.01%E2%80%AFa.%C2%A0m..png)
Características clave de la arquitectura de AlphaFold 3:
- Soporte de entrada multimodal: aminoácidos, nucleótidos, ligandos, iones.
- Predicción estructural conjunta: todas las cadenas y cofactores se predicen juntos.
- Inferencia sin plantillas: predicciones realizadas directamente a partir de la secuencia, guiadas opcionalmente por plantillas.
Conjuntos de datos utilizados en la formación
AlphaFold 3 se capacitó en una amplia colección de datos biológicos, ampliando tanto la secuencia y estructural diversidad utilizada en versiones anteriores.
Bases de datos secuenciales y evolutivas:
El modelo aprovechó varios conjuntos de datos públicos de gran tamaño, incluidos UniProt, BFD y mGnify. Otras fuentes incluyen Uniclust30, RfAM, RNAcentral y la base de datos de nucleótidos.
Bases de datos estructurales:
Se entrenó y evaluó utilizando datos experimentales en 3D del Protein Data Bank (PDB), así como estructuras de ácidos nucleicos de la base de datos de nucleótidos e información de ligandos del Diccionario de componentes químicos (CCD).
Formato y preprocesamiento de datos:
Las funciones de entrada incluyen alineaciones de secuencias múltiples (MSA) y plantillas, aunque el procesamiento de MSA es menos importante que en AlphaFold 2. Las plantillas se recuperan mediante un módulo de búsqueda y las entradas se incrustan en representaciones por pares que procesa el Pairformer, que sustituye al Evoformer AlphaFold 2.
Objetivos de la formación:
AlphaFold 3 se entrenó a través de un marco de difusión generativa, combinando:
- Aprendizaje supervisado: predecir coordenadas atómicas «eliminadas de ruido» a partir de entradas corruptas.
- Destilación cruzada: aumentar los datos con pseudoestructuras de AlphaFold-Multimer v2.3 para mitigar las alucinaciones.
- Aprendizaje generativo sobre datos evolutivos: lo que permite al modelo capturar tanto los pliegues globales como la estereoquímica local en diferentes niveles de ruido.
Estudio de caso: Modelado del complejo CRISPR-Cas9 con AlphaFold 3
Antecedentes
AlphaFold 3 no solo predice el plegamiento tridimensional de las proteínas, sino que también permite a los investigadores explorar cómo los diferentes tipos de biomoléculas interactúan con otras, incluidas las proteínas, el ADN, el ARN y los ligandos pequeños. Para ilustrar esto, hemos realizado dos experimentos de ejemplo con el servidor AlphaFold, que modelaron la hemoglobina humana, un conocido complejo multiproteico, y el sistema CRISPR-Cas9, un ensamblaje proteína-ARN-ADN fundamental para la edición genética moderna.
Antes de profundizar en los resultados, vale la pena explicar brevemente cómo funciona el servidor y cómo interpretar sus métricas de confianza.
Cómo se hacen las predicciones en AlphaFold Server
El Servidor AlphaFold proporciona una interfaz intuitiva para modelar moléculas directamente a partir de sus secuencias.
Cada entidad (proteína, ADN, ARN, ligando o ion) se puede añadir como una cadena separada pegando su secuencia FASTA. Una vez configuradas todas las entidades, el servidor realiza la inferencia mediante el modelo AlphaFold 3, que predice las coordenadas 3D de cada componente de forma simultánea.
%2010.53.36%E2%80%AFa.%C2%A0m..png)
Para reproducir esta configuración, las secuencias utilizadas corresponden a la estructura experimental PDB 5F9R, que contiene la proteína Cas9, el ARN guía y el dúplex de ADN diana.
Los archivos FASTA completos de cada entidad se pueden copiar directamente desde Entrada RCSB PDB 5F9R a través del Página de visualización FASTA.
Los resultados incluyen una estructura pronosticada, puntuaciones de confianza numérica y visualizaciones codificadas por colores que ayudan a evaluar la confiabilidad del modelo de un vistazo.
%2010.54.49%E2%80%AFa.%C2%A0m..png)
El ejemplo muestra una estructura 3D coloreada por pLDDT junto con una matriz de error alineado previsto (PAE). El mapa PAE estima la precisión con la que se colocan las diferentes regiones o cadenas entre sí, el verde oscuro indica un menor error de alineación (mayor confianza) y los tonos más claros indican una mayor incertidumbre en el posicionamiento relativo.
Tras generar una predicción, AlphaFold 3 informa de tres indicadores de confianza principales, que se muestran en la parte superior de cada resultado:
- PLdDT (prueba de diferencia de distancia local prevista) mide la confianza local para cada residuo. Escala de colores: azul = muy alto (> 90), azul claro = seguro (70-90), amarillo = bajo (50-70), naranja = muy bajo (< 50). Estos colores se aplican directamente al modelo 3D para resaltar las regiones rígidas frente a las flexibles.
- PtM (puntuación prevista de modelado de plantillas) evalúa la calidad general del pliegue dentro de una sola cadena.
- IPTM (puntuación de TM pronosticada entre cadenas) cuantifica la confianza con la que AlphaFold 3 predice las interacciones entre diferentes cadenas, como entre subunidades de proteínas o entre una proteína y un ácido nucleico.
Caso 1: Hemoglobina humana: de subunidades a un complejo funcional
La hemoglobina es uno de los conjuntos de proteínas más emblemáticos de la biología. Está compuesta por cuatro subunidades, dos alfa (HBA) y dos beta (HBB), que juntas forman el complejo tetramérico responsable del transporte de oxígeno en los glóbulos rojos. Para explorar cómo AlphaFold 3 maneja tanto las proteínas individuales como las estructuras multiméricas, primero modelamos cada subunidad por separado y luego el complejo de hemoglobina completo que contiene 2 × HBA y 2 × HBB.
Para reproducir este experimento, las secuencias de hemoglobina se obtuvieron de la estructura experimental PDB 13A3N, disponible en Entrada 1A3N de RCSB PDB a través del Enlace FASTA.
%2011.26.39%E2%80%AFa.%C2%A0m..png)
En las tres predicciones, AlphaFold 3 mostró una confianza alta y constante. Tanto las subunidades HBA como las HBB mostraron distribuciones de pLDDT similares, dominadas por regiones de color azul y azul claro que indican una alta confiabilidad local, con solo pequeños extremos flexibles en amarillo o naranja. Las puntuaciones del pTM también fueron altas (0,85 para el HBA y 0,88 para el HBB), lo que confirma la precisión de los pliegues monoméricos. Dado que estas subunidades se modelaron de forma individual, las puntuaciones de la IPTM no son aplicables (no hay interacciones entre cadenas).
Cuando se modeló el tetrámero completo, AlphaFold 3 produjo un resultado aún más fiable, con PtM = 0,89 e iPTM = 0,86, mientras que la estructura prevista parecía casi completamente azul. Esto refleja la mayor certeza del modelo a la hora de predecir la disposición cuaternaria en comparación con las subunidades aisladas.
En términos biológicos, esto tiene sentido, cuando las cadenas alfa y beta se combinan, forman un complejo rígido conservado evolutivamente que AlphaFold puede reconocer a partir de sus datos de entrenamiento, lo que reduce la incertidumbre y aumenta la confianza general.
La estructura resultante se parece mucho al pliegue canónico de la hemoglobina, capturando su organización simétrica y su estabilidad interna.
%2011.27.57%E2%80%AFa.%C2%A0m..png)
La concordancia visual entre la estructura cristalográfica y la predicción de AlphaFold 3 es notable. El modelo reproduce con precisión la orientación relativa de las cuatro subunidades y la cavidad central que caracteriza la forma de unión al oxígeno de la hemoglobina. La alineación entre las dos representaciones destaca cómo AlphaFold 3 no solo captura los detalles a nivel atómico, sino también la geometría global que define el tetrámero funcional. Esta estrecha coincidencia refuerza la fiabilidad de AlphaFold 3 a la hora de predecir conjuntos multiproteicos que tienen arquitecturas bien conservadas en todas las especies.
Caso 2: CRISPR-Cas9: predicción de un complejo proteína-ARN-ADN
Una de las innovaciones más transformadoras introducidas por AlphaFold 3 es su capacidad para modelar no solo proteínas, sino también sus interacciones con otros tipos de biomoléculas, incluidos el ADN, el ARN, los ligandos y los iones. Esto representa un gran avance en comparación con las versiones anteriores, que se limitaban a los complejos proteína-proteína. El modelo ahora captura cómo las proteínas interactúan con los ácidos nucleicos, un aspecto clave de muchos procesos biológicos como la transcripción, la replicación y la edición del genoma.
Un claro ejemplo de esta nueva capacidad multimodal es el sistema CRISPR-Cas9, un complejo molecular que combina una proteína endonucleasa Cas9, un ARN guía (gRNA) y un ADN diana bicatenario. Cas9 utiliza la molécula de ARN como guía para localizar y cortar una secuencia de ADN complementaria, lo que permite una edición genética precisa.
Para probar la capacidad de AlphaFold 3 de predecir ensamblajes tan intrincados, modelamos estas tres entidades simultáneamente en el servidor AlphaFold:
- La proteína Cas9
- El ARN guía
- El ADN bicatenario que contiene el sitio objetivo
(La configuración de estas cuatro entidades se mostró anteriormente en la Figura 6).
%2011.29.06%E2%80%AFa.%C2%A0m..png)
La similitud entre las estructuras experimentales y predichas es sorprendente, especialmente dada la naturaleza multimolecular del sistema. AlphaFold 3 recapitula con precisión la geometría global del ensamblaje del ADN y el ARN de la Cas9, capturando la posición del ARN guía dentro de la hendidura catalítica de la proteína y la curvatura del dúplex de ADN a su alrededor.
Como se ve en el modelo de color PLDDT (panel derecho), la mayor parte de la estructura aparece en azul y azul claro, lo que refleja una fuerte confianza local en Cas9 y la interfaz ARN-ADN. Solo aparecen pequeñas regiones en amarillo o naranja, principalmente dentro de una de las cadenas de ADN (rosa), que también corresponde al área donde se observa la mayor desviación con respecto a la referencia cristalográfica.
Estos resultados destacan cómo AlphaFold 3 se extiende con éxito más allá del plegamiento de proteínas para reconstruir fielmente las interacciones macromoleculares funcionales. Al integrar proteínas y ácidos nucleicos en un único marco predictivo, permite un nuevo nivel de conocimiento de los mecanismos moleculares de la edición del genoma y otros sistemas biológicos complejos.
Aplicaciones biotecnológicas de AlphaFold
AlphaFold está redefiniendo la forma en que los equipos de biotecnología abordan el diseño y el descubrimiento moleculares. Su capacidad para predecir no solo las estructuras proteicas individuales, sino también las interacciones complejas con el ADN, el ARN, los ligandos y otras proteínas, abre la puerta a una amplia gama de aplicaciones en el mundo real.
En el descubrimiento de fármacos, AlphaFold acelera el proceso de identificación de cómo las moléculas terapéuticas se unen a sus objetivos, lo que agiliza el diseño de los leads y reduce la sobrecarga experimental. En lo que respecta a la regulación genética o la replicación viral, el modelo ayuda a visualizar cómo las proteínas interactúan con el ARN y el ADN, lo que aporta nuevos conocimientos sobre los ensamblajes de las RNP y los mecanismos de CRISPR.
Para la biología sintética, permite el diseño de enzimas y complejos multiproteicos completamente nuevos, ampliando los límites de lo que es posible en la ingeniería metabólica o la biocatálisis industrial. En inmunoterapia, AlphaFold desempeña un papel clave a la hora de guiar la modelización de antígenos y anticuerpos, al revelar los puntos de contacto críticos y ayudar a optimizar los anticuerpos monoclonales candidatos.
Y con herramientas como AlphaMisSense, los investigadores pueden combinar las predicciones estructurales de AlphaFold con los datos de las variantes genómicas que ofrecen interpretaciones de las mutaciones y sus consecuencias funcionales.

Conclusión
Desde los primeros enfoques convolucionales de AlphaFold 1 hasta la arquitectura basada en transformadores de AlphaFold 2 y ahora el modelado generativo impulsado por la difusión de AlphaFold 3, la progresión de este sistema refleja la evolución de la propia IA moderna. Cada iteración ha acercado a la comunidad científica a la comprensión de cómo el lenguaje de las secuencias se traduce en la arquitectura de la vida.
Lo que realmente diferencia a AlphaFold 3 es su expansión más allá de las proteínas, su capacidad para predecir las interacciones entre proteínas, ácidos nucleicos, ligandos e iones dentro de un único marco unificado. Este salto hace que el problema pase de predecir pliegues individuales a modelar conjuntos moleculares e interacciones funcionales, como se demuestra en nuestros ejemplos de hemoglobina y CRISPR-Cas9.
Al capturar con precisión no solo los detalles a nivel atómico sino también la interacción dinámica entre las biomoléculas, AlphaFold 3 representa un cambio de la predicción estructural estática a la comprensión mecanicista. Permite a los investigadores visualizar, in silico, complejos a los que antes solo se podía acceder tras años de esfuerzo experimental.
En este sentido, AlphaFold 3 es un testimonio de cómo el aprendizaje automático puede transformar nuestra comprensión de la biología, uniendo la computación y la experimentación para descubrir los principios fundamentales que rigen la vida molecular.
Referencias
[1] Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., Qin, C., Žídek, A., Nelson, A.W.R., Bridgland, A., Penedones, H., Petersen, S., Simonyan, K., Crossan, S., Kohli, P., Jones, D., Silver, D., Kavukcuoglu, K., y Hassabis, D. (2020). Predicción mejorada de la estructura de las proteínas utilizando los potenciales del aprendizaje profundo. Naturaleza, 57, 706-710.
https://doi.org/10.1038/s41586-019-1923-7
(AlphaFold 1: modelo utilizado en CASP13, publicado en 2020.)
[2] Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S.A., Ballard, A., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J.,... Hassabis, D. (2021). Predicción de la estructura de proteínas de alta precisión con AlphaFold. Naturaleza, 596, 583-589.
https://doi.org/10.1038/s41586-021-03819-2
(AlphaFold 2: modelo ganador del CASP14, de código abierto en 2021.)
[3] Abramson, J., Jumper, J., Silver, D., Hassabis, D. y el equipo de DeepMind. (2024). AlphaFold 3 predice la estructura y las interacciones de todas las moléculas de la vida.Naturaleza, 630, 493-500.https://doi.org/10.1038/s41586-024-07487-w
(AlphaFold 3: modelo multimolecular que soporta proteínas, ARN, ADN y ligandos.)


.png)