AlphaFold descoberto: como a IA mapeia os blocos de construção da vida





Introdução
AlphaFold é um sistema revolucionário de IA desenvolvido pela DeepMind que prevê estruturas 3D de proteínas e complexos proteicos de sequências de aminoácidos. Sua evolução do AlphaFold 1 para o AlphaFold 3 redefiniu o campo da biologia estrutural, permitindo que os pesquisadores simulem interações moleculares com precisão sem precedentes. Nesta postagem do blog, rastrearemos a evolução do AlphaFold, exploraremos as arquiteturas de IA e os conjuntos de dados que o alimentam e demonstraremos seu uso na modelagem de interações proteína-RNA-DNA, particularmente com um exemplo biologicamente relevante.
A evolução do AlphaFold: de proteínas únicas a interações complexas
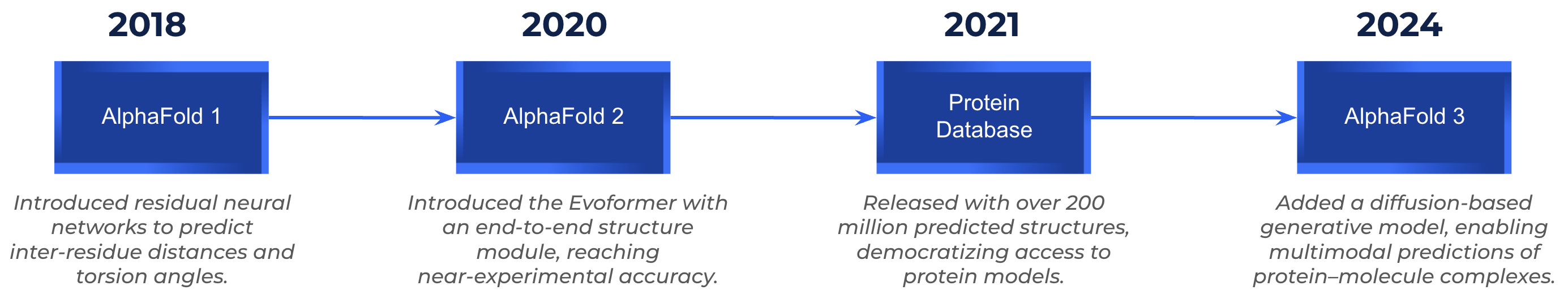
AlphaFold 1 (2018): O avanço no CASP13
Antes do AlphaFold, determinar a estrutura 3D de uma proteína dependia de técnicas experimentais, como cristalografia de raios-X ou microscopia crioeletrônica, que eram altamente precisas, mas também lentas e caras. Para comparar alternativas computacionais, a competição CASP (Avaliação Crítica da Predição de Estrutura) foi criada, desafiando equipes a prever estruturas proteicas invisíveis que seriam posteriormente comparadas com dados experimentais. Em 2018, apesar de décadas de pesquisa, os métodos computacionais ainda lutavam para rivalizar com a precisão experimental. Isso mudou quando o DeepMind entrou em cena.
A primeira versão do AlphaFold ganhou as manchetes em 2018, quando uma equipe de pesquisadores de IA da DeepMind, biólogos estruturais não tradicionais, venceu o desafio CASP13 em sua primeira tentativa. O AlphaFold 1 usou um pipeline de duas partes:
- UM minimização de energia baseada em gradiente descendente processo para montar estruturas 3D a partir das geometrias previstas.
- Um profundo rede neural convolucional residual (CNN) que previu distâncias entre resíduos e ângulos de torção a partir de dados evolutivos.
%2010.40.50%E2%80%AFa.%C2%A0m..png)
Sua principal inovação foi alavancar informações coevolutivas extraídas de vários alinhamentos de sequência (MSAs), que são conjuntos de sequências de proteínas homólogas alinhadas entre as espécies. Usando correlações evolutivas pré-computadas de MSAs, onde certas posições de aminoácidos tendem a sofrer mutações juntas ao longo da evolução, o AlphaFold 1 aprendeu a mapear esses padrões na proximidade espacial 3D entre os resíduos. O modelo produziu um mapa de distância (ou distograma), uma matriz 2D representando as distâncias mais prováveis entre cada par de resíduos, que serviu de base para reconstruir a dobra 3D completa.
%2010.43.20%E2%80%AFa.%C2%A0m..png)
Embora suas previsões nem sempre fossem de nível atômico, elas superaram significativamente os modelos tradicionais de homologia, especialmente na categoria de modelagem livre. O AlphaFold 1 marcou um ponto de inflexão, mostrando que um problema biológico complexo, há muito dominado pela modelagem baseada na física, poderia ser efetivamente reformulado como uma tarefa de aprendizado de máquina.
AlphaFold 2 (2020-2021): Um salto quântico na precisão da previsão
A segunda geração do AlphaFold marcou uma mudança radical na forma como as estruturas das proteínas eram previstas. Enquanto o AlphaFold 1 dependia de recursos pré-computados, como correlações evolutivas, o AlphaFold 2 substituiu esse pipeline de várias etapas por um sistema único de aprendizado profundo de ponta a ponta que aprende diretamente dos dados da sequência. Sua arquitetura foi construída em torno de dois componentes principais:
- Evoformador: uma rede baseada em transformadores que processa simultaneamente informações de vários alinhamentos de sequência (MSAs) e relações de resíduos em pares. Ao criar incorporações internas para o MSA e os pares de resíduos, o Evoformer captura relações evolutivas e espaciais diretamente, sem a necessidade de matrizes de covariância pré-calculadas.
- Módulo de estrutura: uma rede que pega as representações refinadas do Evoformer e prevê as coordenadas atômicas 3D diretamente, refinando-as iterativamente por meio de um processo conhecido como reciclagem, em que cada passagem de previsão melhora a anterior.
Esse design de transformador mudou fundamentalmente a forma como o AlphaFold processava as informações biológicas. Em vez de recursos artesanais, o modelo usa mecanismos de autoatenção, o mesmo conceito por trás de modelos de linguagem natural como o BERT, para capturar dependências de longo alcance entre aminoácidos, tratando a sequência da proteína como uma “frase” em que o contexto é importante para cada resíduo. Melhorias incluídas:
- Aprendizado de ponta a ponta, da sequência bruta à estrutura 3D final.
- Atenção baseada em transformadores que integra informações sequenciais e espaciais.
- Reciclagem de previsões para melhorar a precisão em várias passagens.
AlphaFold 2 venceu o CASP14 por uma ampla margem, alcançando uma precisão quase experimental (GDT > 90) em dois terços das metas. Além da concorrência, seu lançamento de código aberto e a criação do banco de dados de estrutura de proteínas AlphaFold, contendo mais de 200 milhões de estruturas previstas, democratizaram o acesso aos dados de estrutura de proteínas em todo o mundo.
%2010.46.50%E2%80%AFa.%C2%A0m..png)
AlphaFold 3 (2024): Modelando a maquinaria molecular da vida
O AlphaFold 3, lançado em 2024, deu um grande passo à frente ao permitir a previsão de complexos de vários componentes, incluindo:
- Conjuntos proteína-proteína
- Interações proteína-DNA/RNA
- Sítios de ligação proteína-ligante e íon
Ao contrário do AlphaFold 2, que exigia modelos separados para multímeros ou scripts adicionais, o AlphaFold 3 usa um arquitetura unificada para modelar todos os componentes simultaneamente.
Embora o código de inferência do AlphaFold 3 esteja disponível publicamente no GitHub da DeepMind, os pesos do modelo são distribuídos separadamente sob uma licença acadêmica não comercial e devem ser solicitados à DeepMind. Isso torna o AlphaFold 3 uma versão somente para pesquisa, em vez de um modelo totalmente de código aberto, com a maioria dos usuários acessando-o por meio do site oficial Servidor AlphaFold.
O aprendizado de IA por trás do AlphaFold 3
Arquitetura principal: do Evoformer aos modelos de difusão
O AlphaFold 3 mantém o módulo Evoformer para lidar com sequências de entrada, mas substitui o módulo de estrutura por um modelo generativo baseado em difusão. Essa nova abordagem parte de uma nuvem aleatória de átomos e os refina iterativamente em uma estrutura 3D estável, semelhante à de modelos de difusão de imagens, como Difusão estável gere imagens coerentes a partir do ruído.
%2010.50.01%E2%80%AFa.%C2%A0m..png)
Principais características da arquitetura do AlphaFold 3:
- Suporte de entrada multimodal: aminoácidos, nucleotídeos, ligantes, íons.
- Predição estrutural conjunta: todas as cadeias e cofatores previstos juntos.
- Inferência sem modelo: previsões feitas diretamente da sequência, opcionalmente guiadas por modelos.
Conjuntos de dados usados no treinamento
O AlphaFold 3 foi treinado em uma ampla coleção de dados biológicos, expandindo tanto o sequência e estrutural diversidade usada nas versões anteriores.
Bancos de dados sequenciais e evolutivos:
O modelo aproveitou vários grandes conjuntos de dados públicos, incluindo UniProt, BFD e mGnify. Fontes adicionais incluíram Uniclust30, RFam, RNAcentral e o Nucleotide Database.
Bancos de dados estruturais:
Ele foi treinado e avaliado usando dados experimentais 3D do Protein Data Bank (PDB), bem como estruturas de ácido nucléico do Banco de Dados de Nucleotídeos e informações de ligantes do Dicionário de Componentes Químicos (CCD).
Formato de dados e pré-processamento:
Os recursos de entrada incluem vários alinhamentos de sequência (MSAs) e modelos, embora o processamento do MSA seja menos central do que no AlphaFold 2. Os modelos são recuperados por meio de um módulo de pesquisa e as entradas são incorporadas em representações em pares que são processadas pelo Pairformer, que substitui o AlphaFold 2 Evoformer.
Objetivos do treinamento:
O AlphaFold 3 foi treinado por meio de um estrutura de difusão generativa, combinando:
- Aprendizagem supervisionada: prevendo coordenadas atômicas “removidas” a partir de entradas corrompidas.
- Destilação cruzada: aumentando dados com pseudo-estruturas do AlphaFold-multimer v2.3 para mitigar a alucinação.
- Aprendizagem generativa em dados evolutivos: permitindo que o modelo capture dobras globais e estereoquímica local em diferentes níveis de ruído.
Estudo de caso: modelagem do complexo CRISPR-Cas9 com AlphaFold 3
Plano de fundo
O AlphaFold 3 não apenas prevê o dobramento tridimensional de proteínas, mas também permite que os pesquisadores explorem como diferentes tipos de biomoléculas interagem com outras, incluindo proteínas, DNA, RNA e pequenos ligantes. Para ilustrar isso, realizamos dois exemplos de experimentos usando o AlphaFold Server, modelando a hemoglobina humana, um conhecido complexo multiproteico, e o sistema CRISPR-Cas9, um conjunto proteína-RNA-DNA central para a edição moderna de genes.
Antes de mergulhar nos resultados, vale a pena explicar brevemente como o servidor funciona e como interpretar suas métricas de confiança.
Como as previsões são feitas no AlphaFold Server
O Servidor AlphaFold fornece uma interface intuitiva para modelar moléculas diretamente de suas sequências.
Cada entidade (proteína, DNA, RNA, ligante ou íon) pode ser adicionada como uma cadeia separada colando sua sequência FASTA. Depois que todas as entidades são configuradas, o servidor realiza a inferência usando o modelo AlphaFold 3, que prevê as coordenadas 3D de cada componente simultaneamente.
%2010.53.36%E2%80%AFa.%C2%A0m..png)
Para reproduzir essa configuração, as sequências usadas correspondem à estrutura experimental PDB 5F9R, que contém a proteína Cas9, o RNA guia e o duplex de DNA alvo.
Arquivos FASTA completos para cada entidade podem ser copiados diretamente do Entrada RCSB PDB 5F9R por meio do Página de exibição FASTA.
Os resultados incluem uma estrutura prevista, pontuações numéricas de confiança e visualizações codificadas por cores que ajudam a avaliar a confiabilidade do modelo em um piscar de olhos.
%2010.54.49%E2%80%AFa.%C2%A0m..png)
O exemplo mostra uma estrutura 3D colorida por PLddt ao lado de uma matriz de erro alinhado previsto (PAE). O mapa PAE estima a precisão com que regiões ou cadeias diferentes são posicionadas uma em relação à outra, verde escuro indica menor erro de alinhamento (maior confiança) e tons mais claros indicam maior incerteza no posicionamento relativo.
Depois de gerar uma previsão, o AlphaFold 3 relata três indicadores principais de confiança, exibidos na parte superior de cada resultado:
- PLdDT (Teste de diferença de distância local prevista) mede a confiança local para cada resíduo. Escala de cores: azul = muito alto (> 90), azul claro = confiante (70-90), amarelo = baixo (50-70), laranja = muito baixo (< 50). Essas cores são aplicadas diretamente no modelo 3D para destacar regiões rígidas versus flexíveis.
- PtM (pontuação prevista de modelagem de modelo) avalia a qualidade geral da dobra dentro de uma única corrente.
- iPTM (pontuação TM prevista entre cadeias) quantifica com que confiança o AlphaFold 3 prediz interações entre diferentes cadeias, como entre subunidades de proteína ou entre uma proteína e um ácido nucléico.
Caso 1: Hemoglobina humana - das subunidades a um complexo funcional
A hemoglobina é um dos conjuntos proteicos mais emblemáticos da biologia. É composto por quatro subunidades, duas alfa (HBA) e duas beta (HBB), que juntas formam o complexo tetramérico responsável pelo transporte de oxigênio nos glóbulos vermelhos. Para explorar como o AlphaFold 3 lida com proteínas individuais e estruturas multiméricas, primeiro modelamos cada subunidade separadamente e, em seguida, o complexo completo de hemoglobina contendo 2 × HBA e 2 × HBB.
Para reproduzir esse experimento, as sequências de hemoglobina foram obtidas da estrutura experimental. PDB 1A3N, disponível no Entrada RCSB PDB 1A3N por meio do Link FASTA.
%2011.26.39%E2%80%AFa.%C2%A0m..png)
Em todas as três previsões, o AlphaFold 3 exibiu uma confiança consistentemente alta. As subunidades HBA e HBB mostraram distribuições plDDT semelhantes, dominadas por regiões azul e azul claro que indicam alta confiabilidade local, com apenas pequenos terminais flexíveis em amarelo ou laranja. As pontuações de PtM também foram altas (0,85 para HBA e 0,88 para HBB), confirmando dobras monoméricas precisas. Como essas subunidades foram modeladas individualmente, as pontuações do iPTM não são aplicáveis (nenhuma interação entre cadeias está presente).
Quando o tetrâmero completo foi modelado, o AlphaFold 3 produziu um resultado ainda mais confiável, com PtM = 0,89 e iPTM = 0,86, enquanto a estrutura prevista parecia quase inteiramente azul. Isso reflete a maior certeza do modelo em prever o arranjo quaternário em comparação com subunidades isoladas.
Em termos biológicos, isso faz sentido, quando as cadeias alfa e beta se combinam, elas formam um complexo rígido e evolutivamente conservado que o AlphaFold pode reconhecer a partir de seus dados de treinamento, reduzindo a incerteza e aumentando a confiança geral.
A estrutura resultante se assemelha muito à dobra canônica da hemoglobina, capturando sua organização simétrica e estabilidade interna.
%2011.27.57%E2%80%AFa.%C2%A0m..png)
A concordância visual entre a estrutura cristalográfica e a previsão do AlphaFold 3 é notável. O modelo reproduz com precisão a orientação relativa de todas as quatro subunidades e da cavidade central que caracteriza a forma de ligação ao oxigênio da hemoglobina. O alinhamento entre as duas representações destaca como o AlphaFold 3 não apenas captura detalhes em nível atômico, mas também a geometria global que define o tetrâmero funcional. Essa combinação próxima reforça a confiabilidade do AlphaFold 3 na previsão de conjuntos de multiproteínas que têm arquiteturas bem conservadas em todas as espécies.
Caso 2: CRISPR-Cas9: Predição de um complexo proteína-RNA-DNA
Uma das inovações mais transformadoras introduzidas pelo AlphaFold 3 é sua capacidade de modelar não apenas proteínas, mas também suas interações com outros tipos de biomoléculas, incluindo DNA, RNA, ligantes e íons. Isso representa um grande avanço em comparação com as versões anteriores, que eram limitadas aos complexos proteína-proteína. O modelo agora captura como as proteínas interagem com os ácidos nucléicos, um aspecto fundamental de muitos processos biológicos, como transcrição, replicação e edição do genoma.
Um exemplo claro dessa nova capacidade multimodal é o sistema CRISPR-Cas9, um complexo molecular que combina uma proteína endonuclease Cas9, um RNA guia (gRNA) e um alvo de DNA de fita dupla. O Cas9 usa a molécula de RNA como guia para localizar e cortar uma sequência de DNA complementar, permitindo a edição precisa do gene.
Para testar a capacidade do AlphaFold 3 de prever montagens tão complexas, modelamos essas três entidades simultaneamente no AlphaFold Server:
- A proteína Cas9
- O RNA guia
- O DNA de fita dupla contendo o local alvo
(A configuração dessas quatro entidades foi mostrada anteriormente na Figura 6.)
%2011.29.06%E2%80%AFa.%C2%A0m..png)
A semelhança entre as estruturas experimentais e previstas é impressionante, especialmente dada a natureza multimolecular do sistema. O AlphaFold 3 recapitula com precisão a geometria global da montagem Cas9-RNA-DNA, capturando o posicionamento do RNA guia dentro da fenda catalítica da proteína e a curvatura do duplex de DNA ao redor dela.
Conforme visto no modelo colorido PLDDT (painel direito), a maior parte da estrutura aparece em azul e azul claro, refletindo forte confiança local em Cas9 e na interface RNA-DNA. Apenas pequenas regiões aparecem em amarelo ou laranja, principalmente dentro de uma das fitas de DNA (rosa), o que também corresponde à área onde se observa o maior desvio da referência cristalográfica.
Esses resultados destacam como o AlphaFold 3 se estende com sucesso além do enovelamento de proteínas para reconstruir fielmente as interações macromoleculares funcionais. Ao integrar proteínas e ácidos nucléicos em uma única estrutura preditiva, ele permite um novo nível de visão sobre os mecanismos moleculares de edição do genoma e outros sistemas biológicos complexos.
Aplicações biotecnológicas do AlphaFold
A AlphaFold está redefinindo a forma como as equipes de biotecnologia abordam o design e a descoberta moleculares. Sua capacidade de prever não apenas estruturas individuais de proteínas, mas também interações complexas com DNA, RNA, ligantes e outras proteínas, abre uma ampla gama de aplicações no mundo real.
Na descoberta de medicamentos, o AlphaFold acelera o processo de identificação de como as moléculas terapêuticas se ligam aos seus alvos, simplificando o design do eletrodo e reduzindo a sobrecarga experimental. Quando se trata de regulação gênica ou replicação viral, o modelo ajuda a visualizar como as proteínas interagem com o RNA e o DNA, trazendo novos insights sobre os conjuntos RNP e os mecanismos CRISPR.
Para a biologia sintética, ele permite o design de enzimas e complexos multiproteicos totalmente novos, ultrapassando os limites do que é possível em engenharia metabólica ou biocatálise industrial. Na imunoterapia, o AlphaFold desempenha um papel fundamental na orientação da modelagem anticorpo-antígeno, revelando locais de contato críticos e ajudando a otimizar os candidatos a anticorpos monoclonais.
E com ferramentas como o AlphaMissense, os pesquisadores podem combinar as previsões estruturais do AlphaFold com dados de variantes genômicas, oferecendo interpretações das mutações e suas consequências funcionais.

Conclusão
Das primeiras abordagens convolucionais do AlphaFold 1 à arquitetura baseada em transformadores do AlphaFold 2 e agora à modelagem generativa orientada por difusão do AlphaFold 3, a progressão desse sistema reflete a evolução da própria IA moderna. Cada iteração aproximou a comunidade científica da compreensão de como a linguagem das sequências se traduz na arquitetura da vida.
O que realmente diferencia o AlphaFold 3 é sua expansão além das proteínas, sua capacidade de prever interações entre proteínas, ácidos nucléicos, ligantes e íons dentro de uma única estrutura unificada. Esse salto transforma o problema da previsão de dobras individuais em modelagem de montagens moleculares e interações funcionais, conforme demonstrado em nossos exemplos de hemoglobina e CRISPR-Cas9.
Ao capturar com precisão não apenas os detalhes do nível atômico, mas também a interação dinâmica entre as biomoléculas, o AlphaFold 3 representa uma mudança da predição estrutural estática para a compreensão mecanicista. Ele permite que os pesquisadores visualizem, in silico, complexos que antes eram acessíveis apenas por meio de anos de esforço experimental.
Nesse sentido, o AlphaFold 3 é uma prova de como o aprendizado de máquina pode transformar nossa compreensão da biologia, unindo computação e experimento para descobrir os princípios fundamentais que governam a vida molecular.
Referências
[1] Senior, A. W., Evans, R., Jumper, J., Kirkpatrick, J., Sifre, L., Green, T., Qin, C., Žídek, A., Nelson, A. W. R., Bridgland, A., Penedones, H., Petersen, S., Simonyan, K., Crossan, S., Kohli, P., Jones, D. T., Silver, D., Kavavan K. K. e Hassabis, D. (2020). Predição aprimorada da estrutura da proteína usando potenciais de aprendizado profundo. Natureza, 577, 706-710.
https://doi.org/10.1038/s41586-019-1923-7
(AlphaFold 1 — modelo usado no CASP13, publicado em 2020.)
[2] Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A., Ballard, A. J., Cowie, A. Mera-Paredes, B., Nikolov, S., Jain, R., Adler, J.,... Hassabis, D. (2021). Predição altamente precisa da estrutura da proteína com AlphaFold. Natureza, 596, 583-589.
https://doi.org/10.1038/s41586-021-03819-2
(AlphaFold 2 — modelo vencedor do CASP14, de código aberto em 2021.)
[3] Abramson, J., Jumper, J., Silver, D., Hassabis, D. e a equipe DeepMind. (2024). O AlphaFold 3 prevê a estrutura e as interações de todas as moléculas da vida.Natureza, 630, 493-500.https://doi.org/10.1038/s41586-024-07487-w
(AlphaFold 3 — modelo multimolecular que suporta proteínas, RNA, DNA e ligantes.)


.png)